|
We start with the most simple point
defects imaginable and consider an uncharged
vacancy in a simple
crystal with a
base consisting
of only one atomic species - that means mostly metals and semiconductors. |
|
 |
Some call this kind of defect
"Schottky Defect,
although the original Schottky defects were introduced for ionic crystals containing at least two different atoms in the base. |
|
|
We call vacancies and their
"opposites", the self-intersitals,
intrinsic point defects
for starters. Intrinsic simple means that
these point defects can be generated in the ideal world of the ideal crystal.
No external or extrinsic help or stuff is
needed. |
|
To form one vacancy at constant pressure
(the usual situation), we have to add some
free enthalpy GF to the crystal,
or, to use the name commonly employed by the chemical community,
Gibbs energy. |
|
|
GF, the free
enthalpy of vacancy formation, is defined as |
|
|
|
|
|
|
|
|
|
|
|
The index F always
means "formation"; HF thus is the
formation enthalpy of one vacancy, SF the
formation entropy of one vacancy, and
T is always the absolute
temperature. |
|
The formation enthalpy HF in solids is
practically indistinguishable from the formation
energy EF (sometimes written
UF) which has to be used if the volume and not the
pressure is kept constant. |
|
|
The formation entropy, which in elementary
considerations of point defects usually is omitted, must not be confused with
the entropy of mixing or configurational
entropy; the entropy originating from the many possibilities of arranging
many vacancies, but is a property of a
single vacancy resulting from the disorder
introduced into the crystal by changing the vibrational properties of the
neighboring atoms (see ahead). |
|
The next step consists of minimizing
the free enthalpy G
of the complete crystal with respect to the number nV
of the vacancies, or the concentration cV =
nV /N, if the number of vacancies is referred to
the number of atoms N comprising the crystal. We will drop the
index "V" from now now on because this consideration is valid
for all kinds of point defects, not just vacancies. |
|
|
The number or concentration of vacancies in
thermal
equilibrium (which is not necessarily identical to
chemical
equilibrium!) then follows from finding the minimum of G
with respect to n (or c), i.e. |
|
|
|
|
|
∂G
∂n |
= |
∂
∂n |
( |
G0 + G1 +
G2 |
) |
= 0 |
|
|
|
|
|
|
|
with G0 = Gibbs
energy of the perfect crystal, G1 = Work (or energy)
needed to generate n vacancies = n ·
GF, and G2 = – T ·
Sconf with Sconf =
configurational
entropy of n vacancies, or, to use another expression for
the same quantity, the entropy of mixing
n vacancies. |
|
We
note that the partial derivative of G with respect to
n, which should be written as [∂G/∂n]everything else =
const. is, by definition,
the chemical
potential µ of the defects
under consideration. This will become important if we consider chemical
equilibrium of defects in, e.g., ionic
crystals. |
|
The partial derivatives are easily
done, we obtain |
|
|
|
|
|
|
|
|
|
|
|
which finally leads to |
|
|
|
|
|
∂G
∂n |
= |
GF – T · |
∂Sconf
∂n |
= |
0 |
| |
|
|
|
|
|
| |
= |
chemical potential in equilibrium |
|
|
|
|
|
|
We now need to
calculate the configurational entropy Sconf by
using Boltzmann's
famous formula |
|
|
|
|
|
|
|
|
|
With kB = k = Boltzmanns
constant and P = number of different configurations (=
microstates) for the same
macrostate. |
|
|
The exact meaning of P is sometimes a bit
confusing; activate the link
to see why. |
|
A macrostate for our case is any possible combination
of the number n of vacancies and the number N of
atoms of the crystal. We obtain P(n) thus by looking at
the number of possibilities to arrange n vacancies on
N sites. |
|
|
This is a standard situation in
combinatorics; the number we need is given by the
binomial
coefficient; we have
|
|
|
|
|
|
| P |
= |
( |
N
n |
) |
= |
N!
(N – n)! · n! |
|
|
|
|
|
|
|
If you have problems with that, look at
exercise 2.1-1 below. |
|
The calculation of ∂S/∂n now is straight forward in principle,
but analytically only possible with two approximations: |
|
|
1. Mathematical
Approximation: Use the Stirling
formula in
its simplest version for the
factorials, i.e. |
|
|
|
|
|
|
|
|
|
|
|
2. Physical
Approximation: There are always far fewer vacancies than atoms; this
means |
|
|
|
|
|
|
|
|
|
|
As a first result we obtain
"approximately" |
|
|
|
|
|
|
|
|
|
|
If you have any doubts about this
point, you should do the following exercise. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
With n/N =
cV = concentration of vacancies as defined before, we
obtain the familiar formula |
|
|
|
|
|
|
|
|
|
|
|
or, using GF =
HF – T SF |
|
|
|
|
|
| cV |
= exp |
SF
k |
· exp – |
HF
kT |
|
|
|
|
|
|
For self-interstitials, exactly the same
formula applies if we take the formation energy to be now the formation energy
of a self-interstitial. |
|
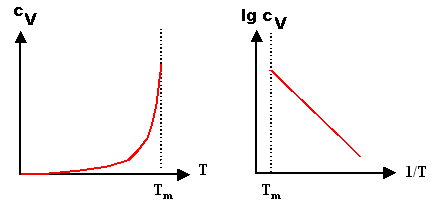
It goes without saying (I hope) that
the way you look at equations like this is via an Arrhenius plot. In the
link you can play with that
and refresh your memory |
|
|
Instead of plotting
cV(T) vs. T directly as in the
left part of the illustration below, you plot the logarithm
lg[cV(T)] vs. 1/T as shown on the
right. |
|
|
In the resulting "Arrhenius plot" or
"Arrhenius diagram" you will get a straight line. The (negative)
slope of this straight line is then "activation" energy of the
process you are looking at (in our case the formation energy of the vacancy),
the y-axis intercept gives directly the pre-exponential
factor. |
|
|
|
|
|
| |
|
|
Compared to simple formulas in
elementary courses, the factor exp(SF/k) might be new.
It will be justified below. |
|
Obtaining this formula by shuffling
all the factorials and so on is is not quite as easy as it looks - lets do a
little fun exercise |
|
|
|
|
|
|
|
| |
|
|
Like always, one can second-guess the
assumptions and approximations: Are they really justified? When do they break
down? |
|
|
The reference enthalpy
G0 of the perfect crystal may not be constant, but
dependent on the chemical environment of the crystal since it is in fact a sum
over chemical potentials including all constituents that may undergo reactions
(including defects) of the system under consideration. The concentration of
oxygen vacancies in oxide crystals may, e.g., depend on the partial pressure of
O2 in the atmosphere the crystal experiences. This is one of
the working principles of Ionics as used for sensors. Chapter
2.4 has more to say to that. |
|
|
The simple equilibrium consideration does not
concern itself with the kinetics of the generation and annihilation of
vacancies and thus makes no statement about the time
required to reach equilibrium. We also must keep in mind that the
addition of the surplus atoms to external or internal surfaces, dislocations,
or other defects while generating vacancies, may introduce additional energy
terms. |
|
|
There may be more than one
possibility for a
vacancy to occupy a lattice site (for
interstitials this
is more obvious). This can be seen as a degeneracy of the energy state, or as
additional degrees of freedom for the combinatorics needed to calculate the
entropy. In general, an additional entropy term has to be introduced. Most
generally we obtain |
|
|
|
|
|
|
|
|
|
|
|
with Zd or
Z0 =
partition functions of
the system with and without defects, respectively. The link (in German) gets
you to a short review of statistical thermodynamics including the partition
function. |
|
Lets look at two examples where this
may be important: |
|
|
The energy state of a vacancy might be
"degenerate", because it is charged and has trapped an electron that
has a spin which could be either up or down - we have two, energetically
identical "versions" of the vacancy and
Zd/Z0 = 2 in this case. |
|
|
A double vacancy in a bcc crystals has
more than one way of sitting at one lattice position. There is a preferred
orientation along <111>, and
Zd/Z0 = 4 in this case. |
| |
|
|
|
The formation entropy is
associated with a single defect, it must not be mixed
up with the entropy of mixing resulting from many defects. |
|
|
It can be seen as the additional entropy or disorder added to the crystal
with every additional vacancy. There is disorder associated with every single
vacancy because the vibration modes of the
atoms are disturbed by defects. |
|
|
Atoms with a vacancy as a neighbour tend to
vibrate with lower frequencies because some bonds, acting as
"springs", are missing. These atoms are therefore less well localized
than the others and thus more "unorderly" than regular atoms. |
|
Entropy residing in
lattice vibrations is nothing new, but quite important outside of defect
considerations, too: |
|
|
Several bcc element crystals are stable
only because of the entropy inherent in
their lattice vibrations. The – TS term in the
free enthalpy then tends to
overcompensate the higher enthalpy associated with non close-packed lattice
structures. At high temperatures we therefore find a tendency for a phase
change converting fcc lattices to bcc lattices which have
"softer springs", lower vibration frequencies and higher entropies.
For details compare Chapter 6 of Haasens
book. |
|
The calculation of the formation entropy, however, is a
bit complicated. But the result of this
calculation is quite simple. Here we give only the essential steps and
approximations. |
|
|
First we describe the crystal as a sum of
harmonic oscillators - i.e. we use the
well-known harmonic approximation. From quantum mechanics we know the energy
E of an harmonic oscillator; for an oscillator number i
and the necessary quantum number n we have |
|
|
|
|
|
| Ei,n |
= |
h ωi
2π |
· (n + 1/2) |
|
|
|
|
|
|
We are
going to derive the entropy from the all-encompassing
partition function of the system and
thus have to find the correct expression. |
|
|
The partition function
Zi of one harmonic
oscillator as defined in statistical mechanics is given by |
|
|
|
|
|
| Z i |
= |
∑
n |
exp – |
h ωi · (n + ½)
2π · kT |
|
|
|
|
|
|
|
The partition function of the crystal then is given by the product of all individual partition function of the
p = 3N oscillators forming a crystal with N
atoms, each of which has three degrees of freedom for oscillations. We
have |
|
|
|
|
|
|
|
|
|
|
From
statistical thermodynamics we know that the free energy F (or,
for solids, in a very good approximation also the free enthalpy
G) of our oscillator ensemble which we take for the crystal is
given by |
|
|
|
|
|
| F = – kT
· ln Z = kT · |
∑
i |
( |
hωi
4πkT |
+ |
ln |
( |
1 – exp – |
hωi
2πkT |
) |
) |
|
|
|
|
|
|
Likewise, the entropy of
the ensemble (for const. volume) is |
|
|
|
|
|
|
|
|
|
|
Differentiating with respect to T yields for the
entropy of our - so far - ideal crystal without defects: |
|
|
|
|
|
| S = k · |
∑
i |
( |
– ln |
( |
1 – exp |
hωi
2π · kT |
) |
+ |
|
) |
|
|
|
|
|
|
Now
we consider a crystal with just one
vacancy. All
eigenfrequencies of all
oscillators change from ωi to a
new as yet undefined value ω'i.
The entropy of vibration now is S'. |
|
|
The formation entropy SF
of our single vacancy now can be defined, it is |
|
|
|
|
|
|
|
|
|
|
|
i.e. the difference in entropy between the
perfect crystal and a crystal with one
vacancy. |
|
It is now time to get
more precise about the ωi, the
frequencies of vibrations. Fortunately, we know some good approximaitons: |
|
|
At temperatures higher then the Debye
temperature, which is the interesting
temperature region if one wants to consider vacancies in reasonable
concentrations, we have |
|
|
|
|
|
hωi
2π |
<< |
kT |
| |
|
|
hω'i
2π |
<< |
kT |
|
|
|
|
|
|
|
which means that we can expand hωi/2π into a
series of which we (as usual) consider only the first term. |
|
Running
through the arithmetic, we obtain as final result, summing over all
eigenfrequencies of the crystal |
|
|
|
|
|
|
|
|
|
|
This now calls for a
little exercise: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For analytical calculations we only
consider next neighbors of a vacancy as contributors to the sum; i.e. we assume
ω = ω´ everywhere else. In a linear approximation, we
consider bonds as linear springs; missing bonds change the frequency in an
easily calculated way. As a result we obtain (for all cases where our
approximations are sound): |
|
|
SF (single vacancy)
» 0.5 k (Cu) to 1.3 k (Au). |
|
|
SF (double vacancy)
» 1.8 k (Cu) to 2.2 k (Au). |
|
These values, obtained by assuming
that only nearest neighbors of a vacancy contribute to the formation entropy,
are quite close to the measured ones. (How formation entropies are measured,
will be covered in chapter 4). Reversing the argumentation, we come to a major
conclusion: |
|
The formation entropy measures the spatial
extension of a vacancy, or, more generally, of a zero-dimensional
defect. The larger SF, the more extended
the defect will be because than more atoms must have changed their vibrations
frequencies. |
|
|
As a rule of thumb (that we justify with a little
exercise below) we have: |
|
|
SF » 1k corresponds to a truly atomic defect,
SF » 10k correponds to
extended defects disturbing a volume of
about 5 - 10 atoms. |
|
|
This is more easily visualized for interstitials
than for vacancies. An "atomic" interstitials can be
"constructed" by taking out one
atom and filling in two atoms without
changing all the other atoms appreciably. An interstitial extended over the
volume of e.g. 10 atoms is formed by taking out 10 atoms and filling in 11 atoms without giving preference in any
way to one of the 11 atoms - you cannot identify a given atom with the
interstitial. |
|
Vacancies or interstitials in
elemental crystal mostly have formation entropies around 1k, i.e. they
are "point like". There is a big exception, however: Si does not fit this picture. |
|
|
While the precise values of formation enthalpies
and entropies of vacancies and interstitials in Si are still
not known with any precision, the
formation entropies are definitely large and probably temperature dependent;
values around 6k - 15k at high temperatures are considered.
Historically, this led Seeger and Chik in 1968 to propose that in
Si the self-interstitial is the dominating point defect and not the
vacancy as in all other (known) elemental crystals. This proposal kicked of a
major scientific storm; the dust has not yet settled. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
So far, we assumed that there is no
interaction between point defects, or that their density is so low that they
"never" meet. But interactions are the rule, for vacancies they are
usually attractive. This is relatively easy to see from basic
considerations. |
|
Let's first look at metals: |
|
|
A vacancy introduces a disturbance in the
otherwise perfectly periodic potential which will be screened by the free
electrons, i.e. by a rearrangement of the electron density around a vacancy.
The formation enthalpy of a vacancy is mostly the energy needed for this
rearrangement; the elastic energy contained in the somewhat changed atom
positions is comparatively small. |
|
|
If you now introduce a second vacancy next to to
the first one, part of the screening is already in place; the free enthalpy
needed to remove the second atom is smaller. |
|
|
In other word: There is a certain binding
enthalpy (but from now on we will call it energy, like everybody else)
between vacancies in metals (order of magnitude: (0,1 - 0,2) eV). |
|
Covalently bonded
crystals |
|
|
The formation energy of a vacancy is mostly
determined by the energy needed to "break" the bonds. Taking away a
second atom means that fewer bonds need to be broken - again there is a
positive binding energy. |
|
Ionic crystals |
|
|
Vacancies are charged, this leads to Coulomb
attraction between vacancies in the cation or anion sublattice, resp., and to
repulsion between vacancies of the same nature. We may have positive and
negative binding energies, and in contrast to the other cases the interaction can be long-range. |
|
The decisive new parameter is the
binding energy E2V
between two vacancies. It can be defined as above, but we also can write down a
kind of "chemical" reaction equation
involving the binding energy E2V (the sign is positive
for attraction): |
|
|
|
|
|
|
|
|
|
|
|
V in this case is
more than an abbreviation, it is the
"chemical symbol" for a vacancy.
|
|
|
If you have some doubts about writing down
chemical reaction equation for "things" that are not atoms, you are
quite right - this needs some special
considerations. But rest assured, the above equation is correct, and you
can work with it exactly as with any reaction equation, i.e. apply reaction
kinetics, the mass action
law, etc. |
|
Now we can do a calculation of the
equilibrium concentration of Divacancies. We
will do this in two ways. |
|
First
Approach: Minimize the total free enthalpy (as before): |
|
|
First we define a few convenient quantities
|
|
|
|
|
|
| GF(2V) |
= |
HF(2V) – TSF(2V) |
|
|
|
|
|
|
|
With Δ S2V =
entropy of association; it is in
the order of 1k - 2k in metals. We obtain in complete analogy to single
vacancies |
|
|
|
|
|
| c2V |
= |
z
2 |
· exp |
S2V
k |
· |
exp – |
HF(2V)
kT |
| c2V |
= |
c1V2 · |
z
2 |
· exp |
ΔS2V
k |
· |
exp |
E2V
kT |
|
|
|
|
|
|
|
The factor z/2 (z
= coordination number = number of (symmetrically identical) next
neighbors) takes into account the different ways of aligning a divacancy on one
point in the lattice as already noticed above.
We have z = 12 for fcc, 8 for bcc and
4 for diamond lattices. |
|
The formula tells us that the
concentration of divacancies in thermal
equilibrium is always much
smaller than the concentration of single vacancies since
cV << 1. "Thermal equilibrium" has
been emphasized, because in non-equilibrium things are
totally different! |
|
|
Some typical values for metals close to their
melting point are |
|
|
|
|
|
| c1V |
= |
10–4 - 10–3 |
| |
|
|
| c2V |
= |
10–6 - 10–5 |
|
|
|
|
|
|
In the second approach, we use the
mass action law. |
|
|
With the reversible reaction 1V + 1V
↔ V2V +
E2V and by using the
mass action law we obtain
|
|
|
|
|
|
(c1V)2
c2V |
= |
K(T) = |
const · exp – |
ΔE
kT |
|
|
|
|
|
|
|
With ΔE
= energy of the forward reaction (you have
to be extremely careful with
sign conventions whenever invoking mass action laws!). This leads to |
|
|
|
|
|
| c2V |
= |
(c1V)2 |
· const–1 |
· exp |
ΔE
kT |
|
|
|
|
|
|
In other words: Besides the
"const.–1" we get the same result, but in an
"easier" way. |
|
|
The only (small) problem is: You have to know
something additional for the determination of reaction constants if you just
use the mass action law. And that it is not necessarily easy - it involves the
concept of the chemical
potential and does not easily account for factors coming from additional
freedoms of orientation. e.g. the factor z/2 in the equation above. |
|
The important point in this context
is that the reaction equation formalism also holds for
non-equilibrium, e.g.
during the cooling of a crystal when there are too many vacancies compared to
equilibrium conditions. In this case we must consider local instead of
global equilibrium, see chapter
2.2.3. |
|
|
© H. Föll